

 Favorite (4)
Favorite (4)
So ein Großprojekt ist die EU-Medical Device Regulation.
Der Hauptzweck dieser Verordnung ist, die Sicherheit von Medizinprodukten zu garantieren, insbesondere auch von Produkten mit neuen technischen Möglichkeiten zur verbesserten Betreuung und Behandlung von Patienten.
Die Verordnung selbst ist in einem außergewöhnlich schnellen Verfahren vom Europäischen Parlament erarbeitet und beschlossen worden. Sie war nach dem Brustimplantate-Skandal in Frankreich notwendig geworden. Hier hatten ganz offensichtlich die Kontrollgremien und -mechanismen versagt. Aus diesem Grund legt die neue Verordnung einen sehr viel höheren Wert auf die Dokumentation aller Prozesse bei der Herstellung von Medizinprodukten. Und außerdem ist die „Kontrolle der Kontrolleure“ verschärft worden.
Gerade vor dem Hintergrund von Fehlentwicklungen in der Vergangenheit und der immer größer werdenden Bedeutung der Medizintechnik war eine Novellierung der alten Regeln erforderlich. Das bestreitet auch niemand. Die Sicherheit von Patienten, aber auch von Ärzten und Pflegern muss immer höchste Priorität haben.
Allerdings wurde bei den neuen organisatorischen Vorschriften an mancher Stelle etwas über das Ziel hinausgeschossen. Der Aufwand für die Beantragung und das Erhalten von Genehmigungen ist sehr hoch und für viele der Klein- und Mittelständischen Unternehmen nicht zu leisten.
Uns schien das ganze erstmal nicht zu betreffen. IVAM ist ein Fachverband für Mikrotechnik: Unsere Mitgliedsunternehmen produzieren zwar seit Jahren Komponenten für Medizingeräte, so wie sie es auch für die Automobilindustrie, das Smart Home oder die Kommunikationstechnik machen. Viele von ihnen waren nicht nach der ISO 13485 zertifiziert.
Auch nach den neuen Regeln sind zwar weiterhin die Inverkehrbringer diejenigen, die genauen Dokumentationspflichten unterliegen. Allerdings haben sich mehrere Dinge geändert, z.B.
Was das genau bedeutet, war vielen zunächst nicht klar. Schnell stellte sich dann aber heraus, dass die Inverkehrbringer nun mehr Daten von ihren Lieferanten benötigen. Und nicht nur einfach Daten: diese sollten natürlich gleich in der Weise aufgearbeitet sein, wie es den Vorschriften der neuen MDR entspricht. Die Komponentenhersteller müssen nun das Know-how aufbauen, um dieser Dokumentationspflicht zu genügen.
Damit stellt sich bei vielen dieser Hersteller die Frage, wie sie mit ihrem geistigen Eigentum umgehen wollen. Soll, darf oder muss der Kunde alle Details wissen? Wer legt fest, was weitergegeben werden muss?
Das zweite o.g. Problem betrifft die Dokumentation der Entwicklung eines Produktes. In vielen Fällen wurden Komponenten, wie beispielsweise Sensoren, ursprünglich für andere Anwendungen entwickelt, z.B. für den Einsatz in einem Haushaltsgerät. Da der gleiche Sensor dann auch als Teil in einem Medizinprodukt Anwendung finden kann, liegen oft die Beschreibungen der ursprünglichen Entwicklungsarbeiten nicht in der geforderten Form vor und müssen nun aufwändig nachgeholt werden.
Audits sollen nach der neuen MDR regelmäßig erfolgen und sollen die Dokumentation der gesamten Lieferkette prüfen. Damit können unangemeldete Audits in Zukunft auch bei den Komponentenherstellern durchgeführt werden, die bisher davon weitestgehend verschont waren. Diese Audits sind ein sehr großer finanzieller und zeitlicher Aufwand. Für alle diese regulatorischen Aufgaben müssen auch kleinere Unternehmen eigene ausgebildete Mitarbeiter haben. Das ist von kleineren Unternehmen nicht mehr leistbar!
Ein viel kritischeres Problem ist allerdings, dass die Auditoren, die „Benannten Stellen“ nun selbst erst noch akkreditiert werden müssen. Diese Akkreditierungsverfahren sind sehr aufwändig und benötigen einen sehr langen Vorlauf. Dies hatte den Effekt, dass sich viele der bisher arbeitenden Benannten Stellen nicht wieder beworben haben. Diejenigen, die weiter machen wollen, müssen sich zunächst intern durch neue Mitarbeiter und Schulungen der bisherigen Kollegen „fit“ machen, bevor sie an die Antragsstellung und in den Akkreditierungsprozess einsteigen könnten.
Zurzeit gibt es noch 58 Benannte Stellen in der EU nach der alten Medizinprodukterichtlinie (MDD), die noch bis zum 26.5.20 arbeiten können. Nach der neuesten NANDO Liste gibt es nur noch zehn Benannte Stellen, darunter BSI in Großbritannien, die nach dem Austritt des Landes aus der EU nicht mehr zertifizieren dürfen und die DEKRA, die sowohl in Deutschland als auch in den Niederlanden arbeiten darf. Damit sind es eigentlich europaweit nur acht Ansprechpartner.
Alle Medizinprodukte, auch diejenigen, die schon jahrelang im Markt erhältlich waren, brauchen nach dem 26.5.20 die Zulassung nach der neuen MDR. Wie soll das gehen, wenn doch gerade erst die ersten Benannten Stellen ihre Arbeit aufgenommen haben?
Als die Dramatik dieses Zustandes so langsam realisiert wurde, kamen auch nach und nach die IVAM-Mitglieder mit Anfragen auf uns zu. Aus regelmäßigen Umfragen wussten wir bereits, dass viele Unternehmen unter den europäischen Regularien in vielen Bereichen leiden mussten.
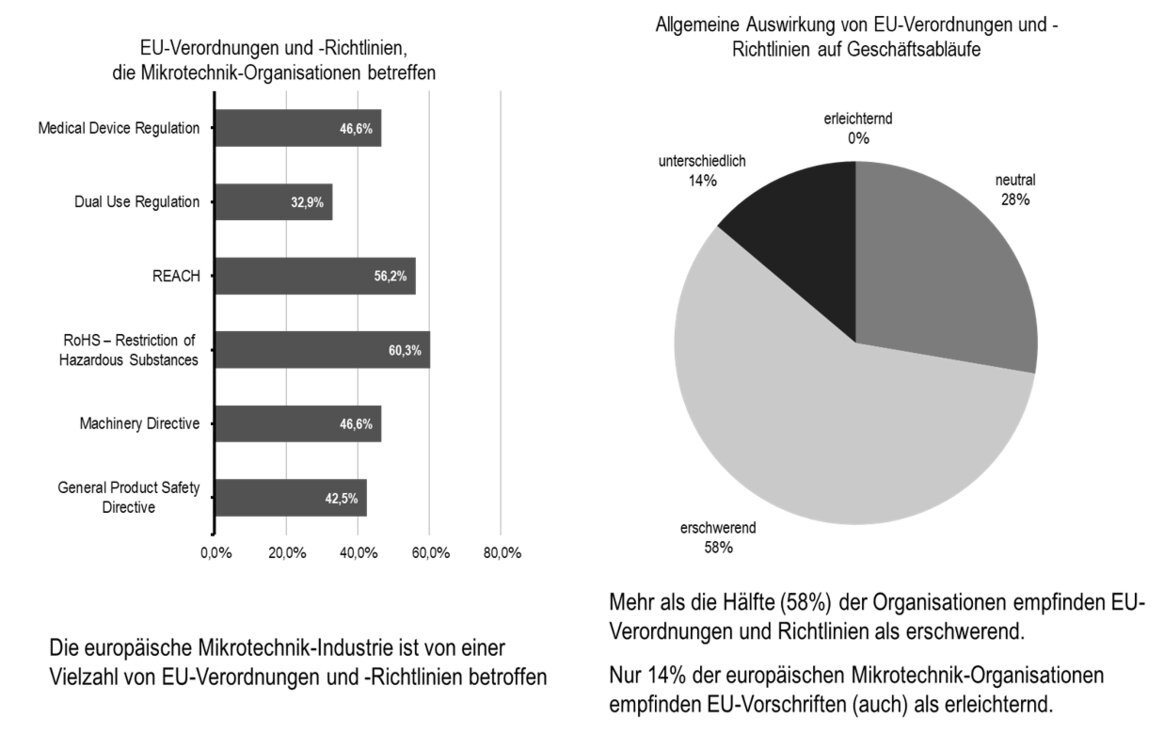
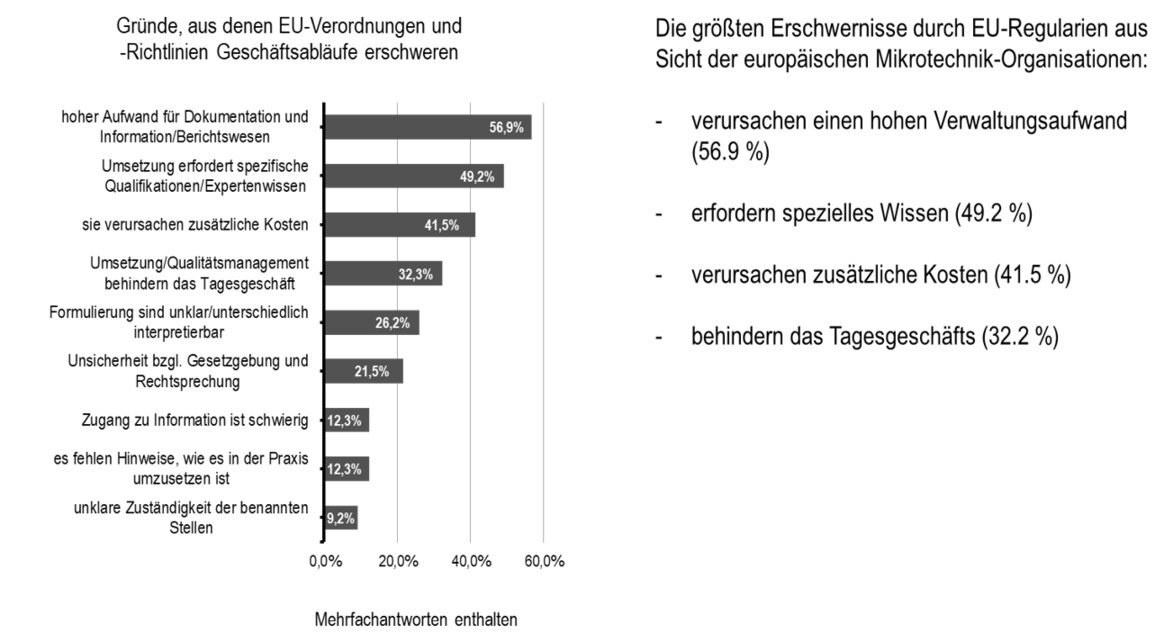
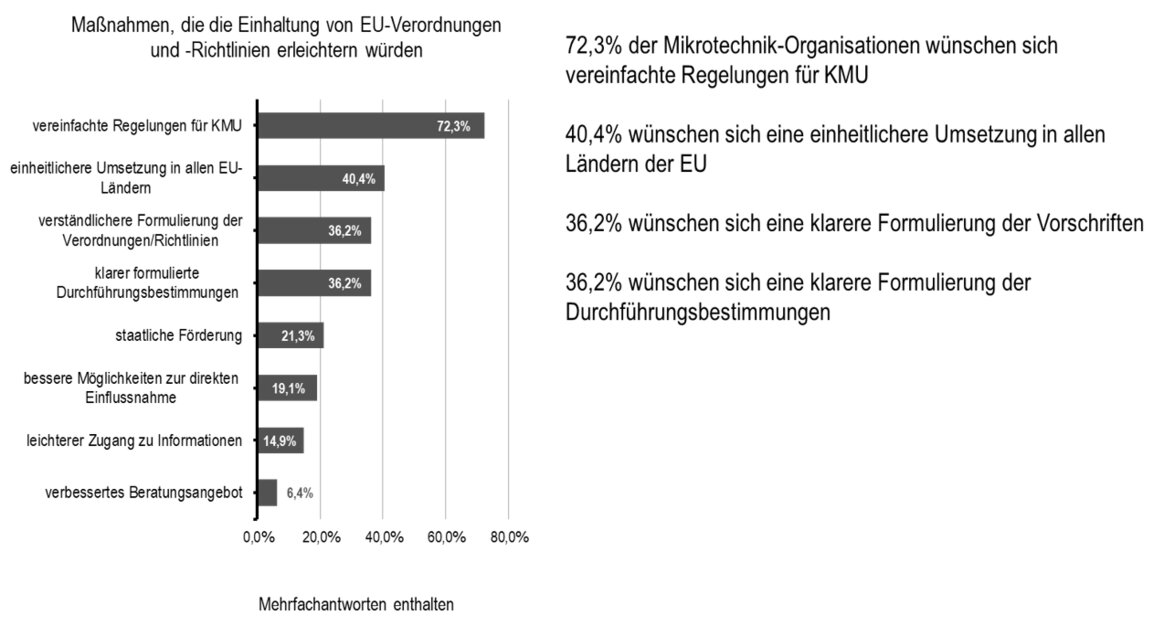
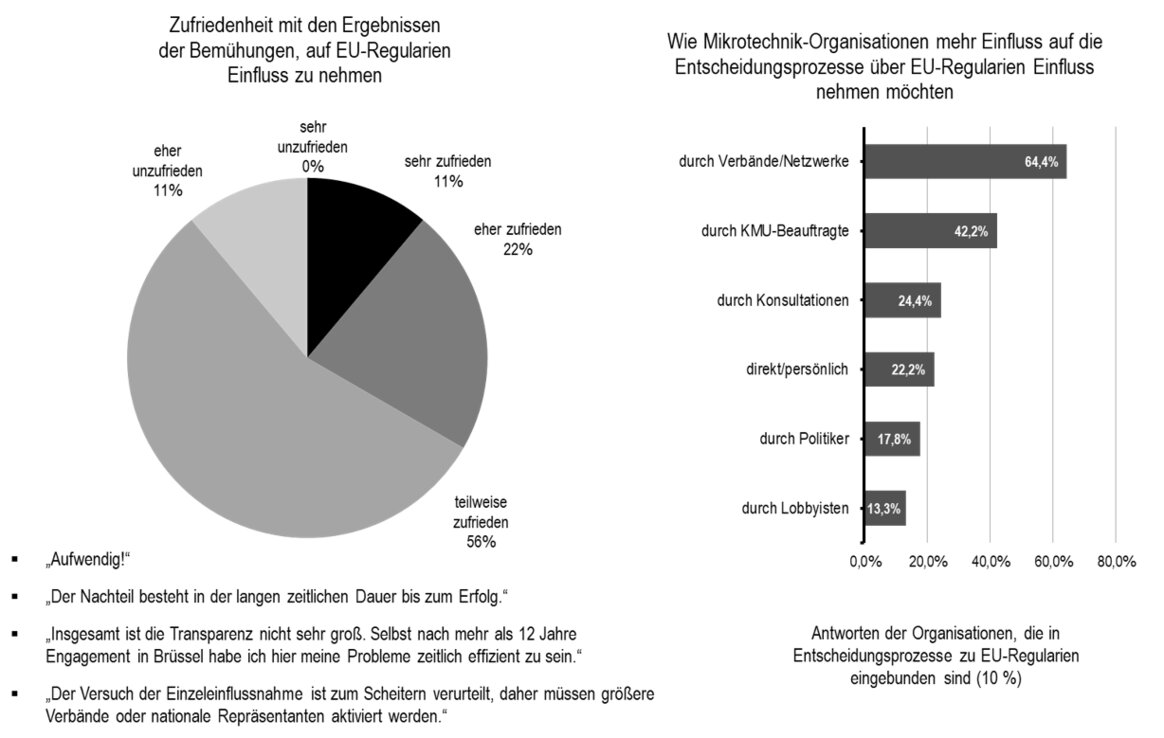
Alle Unternehmen, die uns jetzt wegen der Einführung der neuen MDR, kontaktiert hatten, hatten hauptsächlich ein Informationsdefizit:
Einige Firmen hatten sich aufgrund der aktuellen Lage bereits entschlossen, ihre Medizintechniksparte aufzugeben! Das wäre eine Katastrophe für die Medizintechnik und den deutschen Hightech-Standort. Noch immer kommen eine große Zahl von Innovationen aus dem Mittelstand, der unter der MDR besonders leiden muss.
In welcher Weise kann nun ein Mikrotechnikverband seinen Mitgliedern bei einem solchen Thema helfen?
Wir können erstens dafür sorgen, dass unsere Mitglieder über alle notwendigen Informationen verfügen. Zweitens können wir versuchen, die Rahmenbedingungen zu verbessern. In beiden Bereichen ist es sinnvoll, sich mit Partnern auszutauschen und gemeinsame Aktivitäten zu starten.
Ganz wichtig sind dabei die Zusammenarbeit mit weiteren Verbänden, die sich auf Medizintechnik spezialisiert haben. Wir sind in regelmäßigem Austausch z.B. mit dem BVMed, Medical Mountains, Forum MedTech Pharma und anderen. Hier bekommen wir wichtigen Input für unsere Verbandsaktivitäten und wir können unsere Mitglieder bei Fragen auch dorthin vermitteln.
Bei verschiedenen Informationsveranstaltungen haben wir darüber hinaus auch eine Reihe von Experten aus Benannten Stellen und Beratungsunternehmen eingeladen, die dann konkrete Fragen der Mitglieder beantworten konnten.

Sehr wichtig war uns aber auch die Verbesserung von Rahmenbedingungen und insbesondere die Sensibilisierung der politisch Verantwortlichen in den Bundesländern, beim Bund und in der EU. Bei einer solchen politischen Lobbyarbeit ist der Erfolg an die Größe gekoppelt. Aus diesem Grund waren wir dann auch sehr froh, dass wir mit dem Mikrotechnik-Verband microTEC Südwest einen guten Partner für diese Aktivitäten gefunden hatten.
Auf unserer Agenda standen ein paar Forderungen, die unseren Mitgliedsunternehmen den Übergang in die neue MDR erleichtern sollten. Ziel dabei war, das Informationsdefizit bei unseren Mitgliedern zu verkleinern, ihnen damit den Übergang planbarer und berechenbarer zu machen, und sie von einem Ausstieg aus der Medizintechnik abzuhalten.
Unsere Forderungen haben wir gleich im Jahr 2017 formuliert:
Der letzte Punkt hat gar nicht direkt mit der MDR zu tun. Aber bei den Recherchen zu unseren Aktivitäten ist immer wieder aufgefallen, dass in den Entscheidungsgremien des deutschen Gesundheitssystems Technologiefirmen nur unzureichend vertreten sind und deshalb auch nicht bei z.B. Gesetzesvorhaben gehört werden.
Zum Beispiel entscheidet der Gemeinsame Bundesausschuss regelmäßig über Kostenerstattung von Leistungen mit Medizingeräten. Aber kein Medizingerätehersteller-Verband ist dort vertreten. Immerhin hat z.B. der BVMed ein Stellungnahmerecht – hinterher, wenn die Beschlüsse im Prinzip gefasst worden sind. Wir unterstützen deshalb sehr den Wunsch des BVMed, einen permanenten Sitz in diesem Gremium zu erhalten.
Am 14.6.2018 konnten IVAM und microTEC Südwest ein parlamentarisches Frühstück in Berlin organisieren, bei dem neben Vertretern aus den zuständigen Landesministerien (NRW und BW) auch Mitglieder des Bundestagsgesundheitsausschusses und die Bundesministerien für Gesundheit, Forschung und Wirtschaft involviert waren. Zusammen mit dem Geschäftsführer des BVMed, Herrn Schmitt, konnten die Geschäftsführer und Vorstände von IVAM und microTEC Südwest die Dringlichkeit der Lage noch einmal vorstellen und diskutieren.
In den Diskussionen waren sich eigentlich alle über die Problematik, insbesondere für KMU, einig. Allerdings kam bei der Diskussionen über Verbesserungsvorschläge immer wieder die Aussage: „Da kann man nichts machen. Das hat die EU beschlossen. Das kann nicht geändert werden.“
Mit microTEC Südwest zusammen hatten wir dann aber im Nachgang unsere Befürchtungen in einer Pressemeldung zusammengefasst:
Unsere konkreten Forderungen waren insbesondere:
Ich würde jetzt nicht behaupten wollen, dass alle in den letzten beiden Jahren erreichten und umgesetzten Maßnahmen wegen IVAM erfolgt sind. Aber ich bin überzeugt, dass wir im Zusammenspiel aller Akteure unseren kleinen Beitrag dazu geleistet haben.
Nach unseren Veranstaltungen fanden diverse Telefonate und Meetings mit Vertretern aus den Landesregierungen NRW und BW statt. Danach fand eine Sitzung der Landeswirtschaftsminister statt. Die Beschlüsse der Konferenz vom 27. und 28. Juni 2018 zum Tagesordnungspunkt „Transfer- und Markteintrittsförderung von Innovationen in der Medizintechnik“ haben wir umfänglich begrüßt.
Parallel dazu haben wir inhaltlichen Input für eine "Kleine Anfrage" im Bundestag an die Bundesregierung gegeben. Diese Kleine Anfrage wurde von der Abgeordneten Helling-Plahr und der FDP-Fraktion eingebracht.
Es gab 42 Fragen unter Zuarbeit von IVAM, z.B. zu Fristverlängerungen, Minimierung des Aufwands von KMU, Akkreditierung von Benannten Stellen.
Die Bundesregierung antwortete relativ schnell. Sie bestätigte darin unsere Befürchtungen, dass die Verordnung nicht fristgerecht zum 26. Mai 2020 umgesetzt werden kann. Bundesgesundheitsminister Spahn will bei der EU eine Lösung herbeiführen.
Am 25. November 2019 beschloss die EU dann eine „Grace Period“: Alle Produkte der Klasse I, die bis zum 26 Mai 2020 eine Zertifizierung nach den alten Regularien erhalten haben, dürfen weiterhin bis Mai 2024 in Verkehr gebracht werden!
Das entlastet die wenigen Benannten Stellen, die sich nun um die neuen Produkte kümmern können. Damit war die wichtigste Forderung der Medizintechnik-Hersteller zumindest teilweise erfüllt.
Die Landesregierung Baden-Württemberg hat ein Programm aufgelegt, das von BIOPRO verwaltet wird und uns von Frau Ref in einem Vortrag auf der Informationsveranstaltung von IVAM und microTEC Südwest bei Roche in Mannheim vorgestellt wurde.
Die Landesregierung unterstützt dabei die Erarbeitung von MDR-Unterlagen für spezifische Produktgruppen. Hierbei werden keine Einzelfirmen unterstützt, sondern jeweils eine Gruppe von Unternehmen, die sich für diese Produkte interessiert.
Damit ist Baden-Württemberg ein Vorreiter. Wir hoffen, dass andere Bundesländer diesem Beispiel folgen, um ihren jeweiligen Unternehmen die gleichen Chancen bei der Umsetzung der MDR zu ermöglichen.
Wie oben schon erwähnt, starten die Dokumentationspflichten bereits bei den ersten Entwicklungsschritten für ein Projekt. Es hatte uns insbesondere erstaunt, dass Forschungseinrichtungen sich nicht bewusst waren, dass sie damit ebenfalls die Unterlagen zu den Forschungsergebnissen von Produkten erstellen müssen, die später in Medizingeräte kommen sollen.

Wir hatten deshalb am 15. November 2018 auf dem COMPAMED HIGH-TECH Forum by IVAM eine gemeinsame Infoveranstaltung für Forschungseinrichtungen zur EU-MDR organisiert, zusammen mit microTEC Südwest und Medical Mountains.
Inzwischen haben viele der Forschungseinrichtungen eigene MDR-Beauftragte und können so die KMU bei gemeinsamen Entwicklungsprojekten unterstützen.
Darüber hinaus bestätigte das Bundesforschungsministerium, dass Kosten für die Erstellung von MDR-konformen Unterlagen und Prozessen in Forschungsprojekten förderfähig sind.
Am 31.01.2020 kamen mehr als 30 Mitglieder von IVAM und microTEC Südwest sowie Interessierte aus der Industrie beim Gastgeber Roche Diagnostics GmbH in Mannheim zusammen, um unter dem Motto „Der Countdown zur MDR läuft!“ über die Medical Device Regulation (MDR) zu informieren und sich über die Herausforderungen auszutauschen.

Die wesentlichen Ziele bestanden darin umfassend zu informieren, praktische Anregungen zum Umgang mit der MDR zu geben und einen offenen Erfahrungsaustausch zu ermöglichen. Dazu hatten wir MDR-Experten eingeladen, die die Thematik aus verschiedensten Perspektiven beleuchteten.

Drei Vorträge informierten über die geltenden Regelungen
So erläuterte Michael Bothe von der DQS Medizinprodukte GmbH die Pflichten der Hersteller, die durch die MDR entstehen. Dabei betonte er, dass es für Komponentenhersteller kein separates Konformitätsbewertungsverfahren gibt. Die Hersteller des Medizinprodukts müssen somit beim Komponentenhersteller Qualitätssicherungsmaßnahmen einfordern. Insgesamt muss aus seiner Sicht auf die technische Dokumentation sowie das sorgfältige Risikomanagement ein besonderes Augenmerk gelegt werden.
Michael Maier von Medidee Services sieht hinsichtlich der MDR für In-vitro-Diagnostika (IVDR) noch einiges auf die Unternehmen zukommen. Er empfiehlt, die Forschung und Entwicklung frühzeitig einzubinden und jetzt eine Bereinigung des Produktportfolios vorzunehmen. Ein weiterer wichtiger Aspekt der MDR bestehe in der Klassifizierung der Produkte.
Dr. Alexander Theis von der Scientific Consulting Company berichtete, dass viele Produkte durch die MDR in höhere Klassen umklassifiziert werden müssen, was zu einem höherem Aufwand für die Unternehmen führt.
Neben den regulatorischen Aspekten wurden auch Lösungswege aufgezeigt. Wie bereits erwähnt, beschreitet das Land Baden-Württemberg derzeit mit einem MDR-Soforthilfe-Programm neue Wege und übernimmt damit eine Vorreiterfunktion. Die Maßnahmen des Programms zur Unterstützung der Medizintechnikunternehmen stellte Caroline Ref von der BIOPRO Baden-Württemberg vor.
Antje Nagler von Roche Diagnostics führte eindrucksvoll aus, wie Roche den Herausforderungen der MDR und IVDR begegnet. Als Hauptherausforderungen beschrieb sie den Umgang mit dem Interpretationsspielraum und die mangelnde Verfügbarkeit benannter Stellen.
Aus Perspektive eines Start-ups schilderte Dr. Michael Lauk, die Vorteile darin, als unbeschriebenes Blatt in die Regulatorik einzusteigen und so die Prozesse zu erlernen. Nachholbedarf sieht er allerdings bei der Qualifikation von Experten.
Bei der abschließende Podiumsdiskussion konnte mit den Experten von BVMed Bundesverband Medizintechnologie e.V., IVAM, microTEC Südwest und DQS Medizinprodukte diskutiert werden. Dabei ging es unter anderem um den Einfluss der MDR auf den deutschen Markt, die Frage, wie Audits in der Zukunft durchgeführt werden oder wie die technische Dokumentation auszusehen hat.
Die neue EU-MDR stellt insbesondere die Komponentenhersteller (KMU) vor große Herausforderungen, aber:
Die Arbeiten nach der neuen MDR bleiben schwierig, zeitaufwändig und kostenintensiv! Aber sie sind machbar! Wenn Sie Fragen zur MDR haben oder über Ihre Erfahrungen berichten wollen, dann können Sie sich gern an uns wenden!
